コンピュータはゲノムの理解に不可欠です。
[知識情報処理]
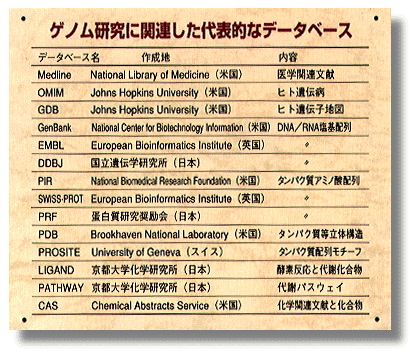
ヒトゲノム計画を始めるにあたって、従来の生物科学が直面したことのない非常に大きな問題があることが分かっていました。それは、計画が進めば進む程、扱うデータの量が膨大なものになるということです。もう一度新聞の例を挙げて考えてみましょう。数10年分の新聞の山があったとします。その中で『一番最初に「ゲノム」という言葉が登場したのは何年何月何日の新聞ですか?』という問いに対して、手作業で1ページづつ見ていくのはほとんど不可能です。しかし、すべての新聞の記事がデータベース化されていていれば、事情はまったく違います。検索ソフトを用いて「ゲノム」という言葉をさがせば、すぐに答えが出てきます。割合単純なデータである新聞のテキストの場合でも、コンピュータの威力は大変なものですが、ヒトゲノム計画の場合はさらにデータは複雑です。まず、配列のデータだけを考えてみても、二つの配列が完全に一致することはなく類似度が問題となります。どのくらい似ているか、またどこが似ているかということなどを定量的に解析することは、コンピュータでも非常に時間がかかります。しかし、類似度の解析からは、生物種同士の進化的な近さを評価することができますし、たんぱく質の機能の推定も可能となります。それだけではなく、データベースの種類はが一つではなく、遺伝子の地図、DNAの文字配列、アミノ酸配列、タンパク質の立体構造、遺伝子と遺伝子の関係、遺伝病、これらに関係する文献など様々なデータベースがあります。このように複数のデータベースがあり、しかもデータがそれぞれお互いに非常に関係が深いと、単に一つのデータベースの中だけで検索するのではなく、データベースからデータベースへわたり歩いて総合的なデータを収集することが求められます。例えば、ある遺伝病に関心があったとすると、まず遺伝病のデータベースを検索します。その遺伝子が見付かれば、そのDNA文字配列を探すためにDNA塩基配列データベースを探しに行きます。さらにそれに関係するアミノ酸配列や、タンパク質の立体構造のデータベースを見てみたり、文献を検索して付加的な情報を探すということになるでしょう。このように、複雑な検索ができるような統合化されたデータベースが、ゲノム研究の大きな流れとなっています。
しかし、ゲノム研究における知識情報処理は、データを蓄え、検索するということにとどまりません。ゲノム研究の情報処理には、まったく質的に違う側面があります。一つは、生物のシステムがあまりにも複雑だということであり、他方はシステムを構成する機能素子(すなわちタンパク質)の機能のメカニズムをまだ私たちは理解していないということです。ある生物種(たとえばヒト)のゲノムを完全に解読したとき、数千ないし数万の遺伝子が提示されますが、その何割かは全く機能の分からないたんぱく質に対応しています。現在は、まだそのようなデータに対する情報処理が十分確立していませんが、将来研究が進めば、次のような情報処理が行われることになります。それらのたんぱく質のアミノ酸配列がどのような立体構造を作り上げるかが予測されます。次に、すべての機能単位の組み合わせとして、細胞、組織、個体の形や挙動をシミュレーションすることになるでしょう。将来はシステムとしての生物をコンピュータの中で構築することができるようになるはずです。